Getting Input Data
Downloading input files for OrthoFinder
Let’s imagine we want to perform a phylogenomic analysis across a set of species:
Dingo (Canis lupus dingo), Great spotted kiwi (Apteryx haastii), Kakapo (Strigops
habroptila), Platypus (Ornithorhynchus anatinus), Tammar wallaby (Notamacropus
eugenii), and Common wombat (Vombatus ursinus).
OrthoFinder requires as input the amino acid sequences for all the protein coding genes in your species of interest. In this section of the tutorial we will discuss how to get these files for the species that you want to analyse. We will cover three major websites for getting this data, Ensembl, NCBI, and Phytozome.
-
Ensembl
Recommended if you want to analyse eukaryotic species, especially vertebrates and model organisms. -
NCBI
Recommended if you want to analyse prokaryotic species. -
Phytozome
Recommended if you want to analyse plants.
A key consideration when getting input data for OrthoFinder is gene transcripts. When you download the amino acid sequences for a genome, you might have multiple sequences for the same gene. This is because a single gene can produce multiple transcripts, which each might produce a different protein. You can see an example of transcripts in this table of the Actin gene on Ensembl.
If we ran OrthoFinder on these raw files it would take much longer than necessary, and could lower the accuracy. We therefore want to extract just the longest transcript variant for each gene. OrthoFinder provides scripts to do this, which we will learn how to use later.
Getting data from Ensembl
First, we go to the Ensembl webpage with the list of species. You can also use the new beta website, which is updated more regularly.
We can then search for our first species (Canis lupus dingo), and click on the
link This will take us to the ensembl page for that species
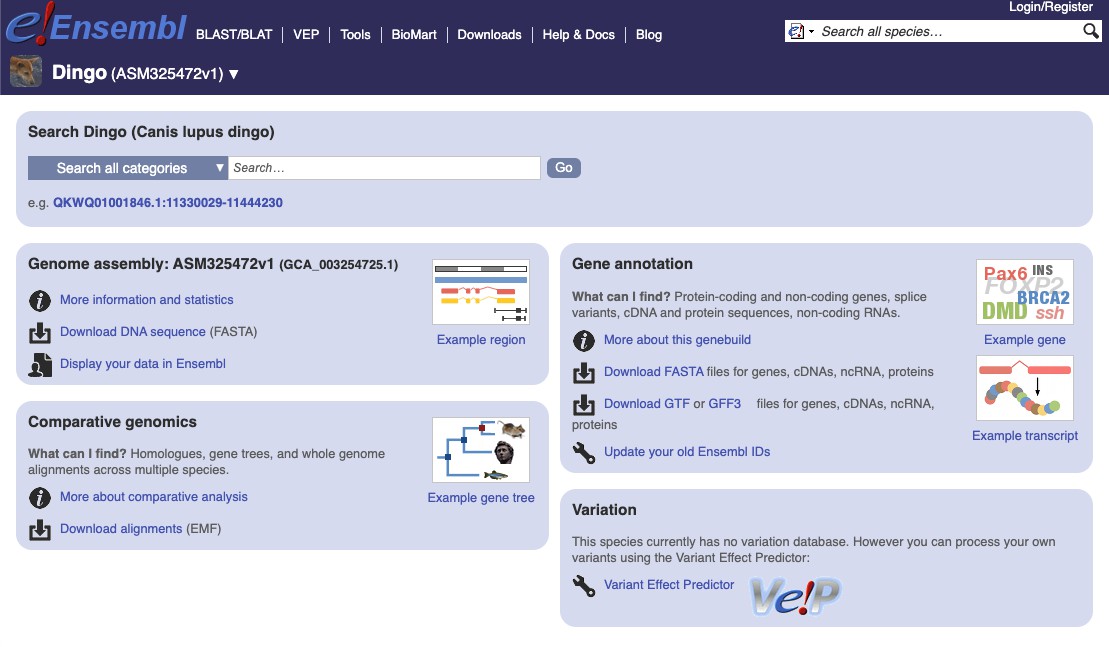
On the right hand side under the heading Gene annotation we can press the link to
Download FASTA files for the genome.
This will take us to a webpage with some folders.
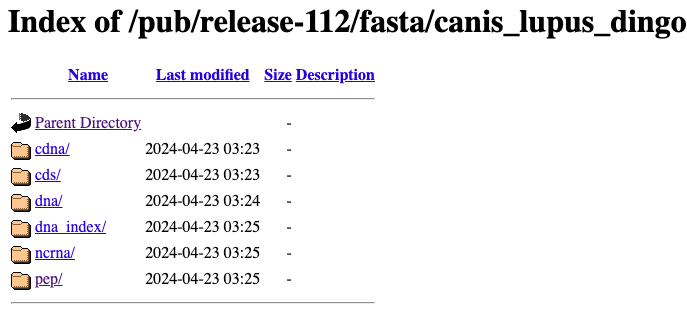
We need to click on the pep/ folder, and then click on the file that ends in .pep.all.fa.gz
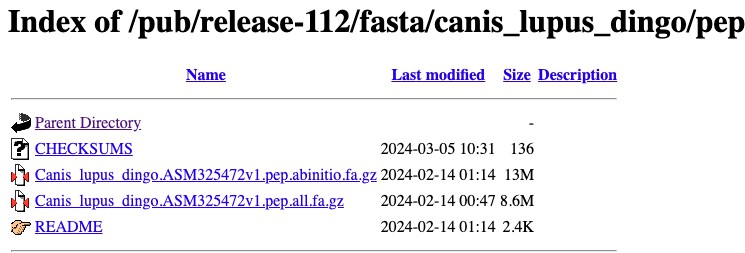
We can then repeat this step for our other species, and place the files that are
downloaded into a folder on our computer. I have named my folder Proteomes.
The files are stored as .gz compressed files to save space, but we now need to expand the files so that we can access the sequences. You can either double-click on them all, or use the command line
gunzip *.gz
We’ll use a script provided with OrthoFinder to extract just the longest transcript variant per gene and run OrthoFinder on these files:
You can find the script here
To run the script, first place it in the Proteomes folder. Then, open the command line and navigate to the Proteomes folder (using cd). You can then use the following command to run the script
for f in *fa ; do python primary_transcript.py $f ; done
The script will generate a new folder called primary_transcripts, which contains our files.
Shortening the filenames is a good idea as it keeps the results tidy as the filenames are
used to refer to the species, e.g. I shortened it to Canis_lupus_dingo.fa.
Our data is now ready for OrthoFinder. You can go to the ‘Beginner Tutorial’ to see how to run OrthoFinder, or check out the other guides on getting input data from other sources
Getting data from NCBI
We start by going to https://www.ncbi.nlm.nih.gov/ and searching for our first species
We want to find the genome of this species, so scroll down to the Genomes section,
and click on Assembly / Genome
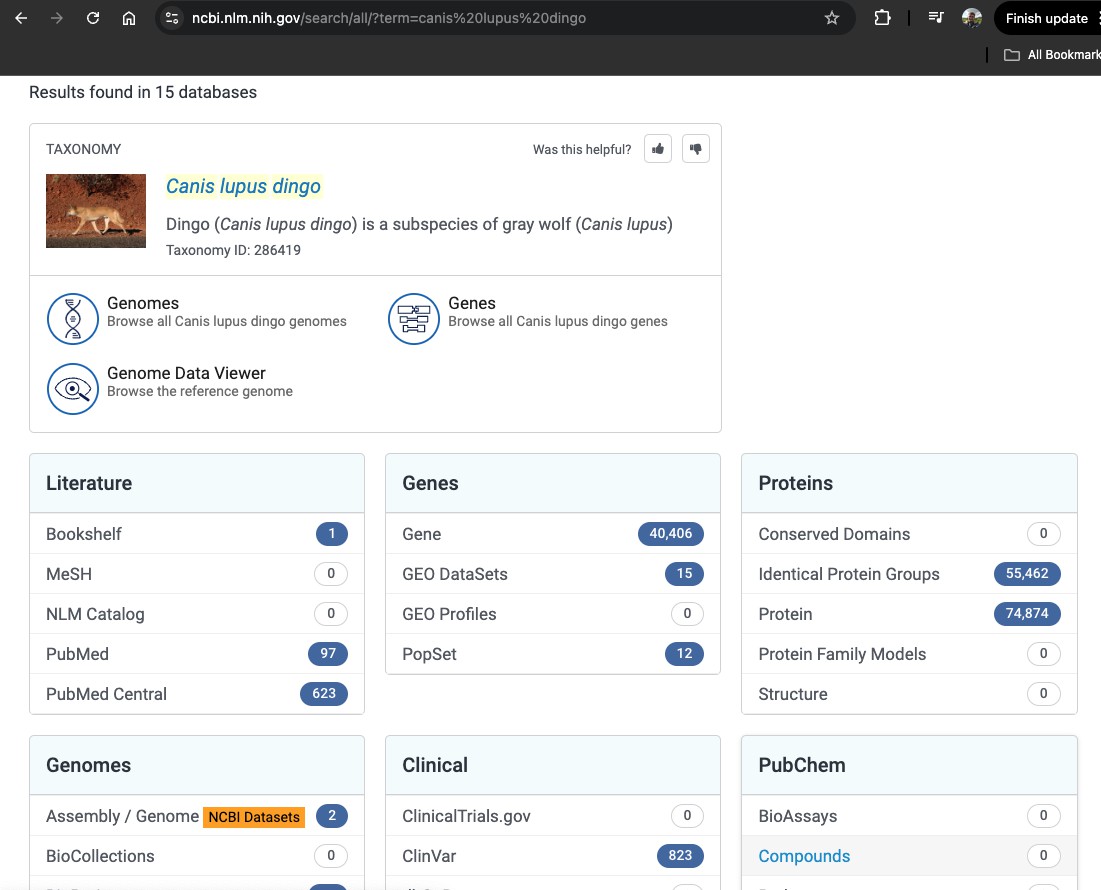
There might be several genomes listed on this page. We are going to click on the one with the green tick, which shows us the reference genome.
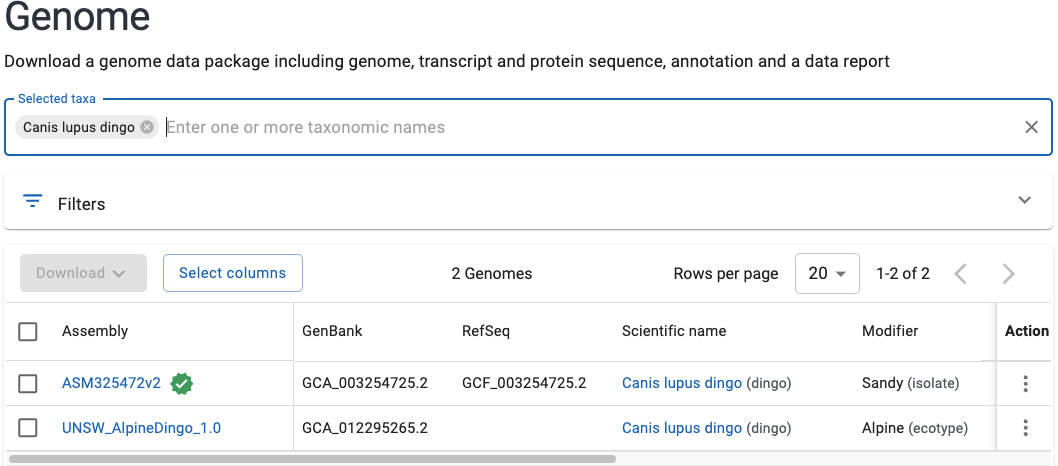
We can then click the blue Download button, and click the boxes to select the curated
RefSeq only annotation, and that we want the GFF and protein FASTA files. We can
also give our download a useful name (such as the name of the species).
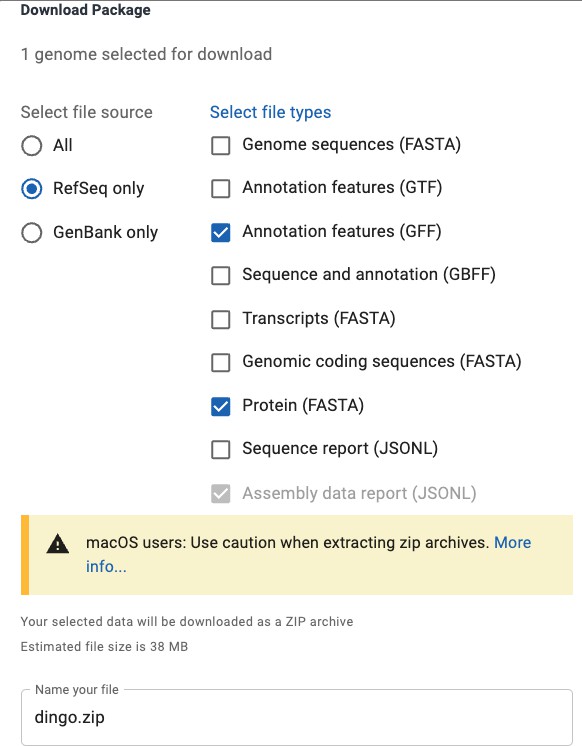
We can then click the Download button, which downloads a zip file.
Repeat this step for all of the species that you want to use. For some species in our example list, like the Kiwi, there is no RefSeq genome with protein fasta available. For these species, it is recommended to use Ensembl.
Now, we need to deal with the issue of multiple transcripts per gene. For our first
species, the Dingo, there are about 20,000 genes. However, if we look at the
protein.faa file that we have downloaded, it has about 75,000 sequences.
We can use the script ncbi_primary_transcript.py to extract the longest
transcript per gene.
Place the script in the folder that has the zip folders, and run the following line of code in the command line
python ncbi_primary_transcripts.py
This will then make a folder named primary_transcripts, which is ready to run
Orthofinder on.
If you want to download data in bulk, you can also use the NCBI Datasets tool, which can run on the command line. For more info, see here
Getting data from Phytozome
First, go to the Phytozome data portal website. Here, you can search for a species, such as Oryza sativa
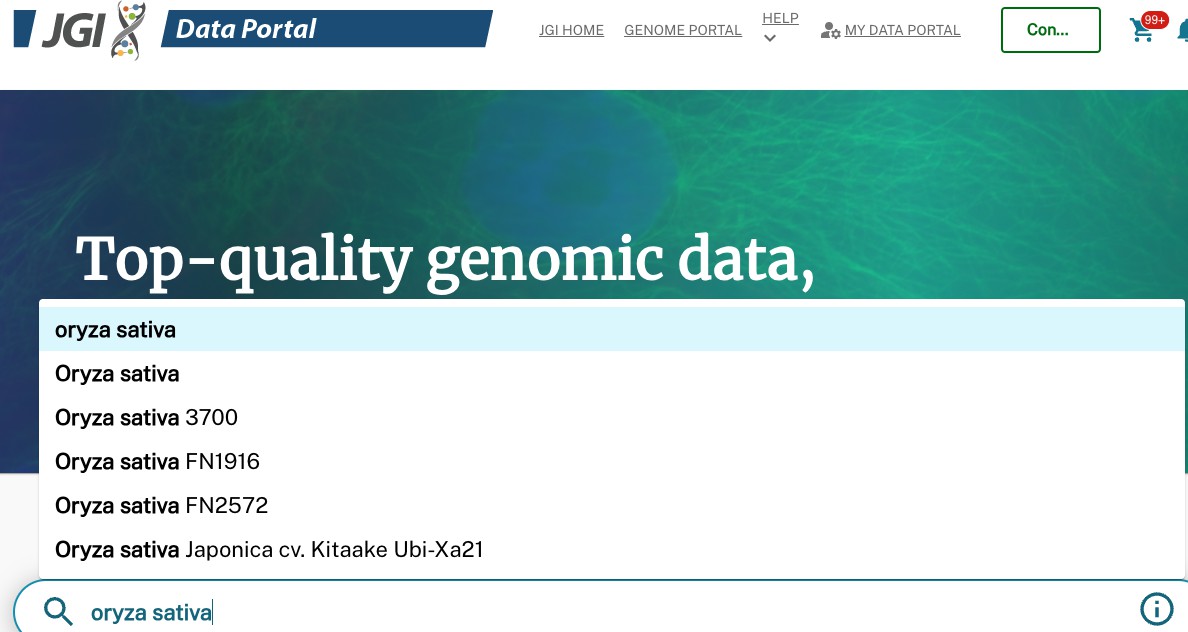
You can then find the genome that want to download, and click on it
The file that we want ends in primaryTranscriptOnly.fa.gz. Luckily for us, Phytozome
already deals with the problem of multiple transcripts. We can select that file, and click
Add to Cart.
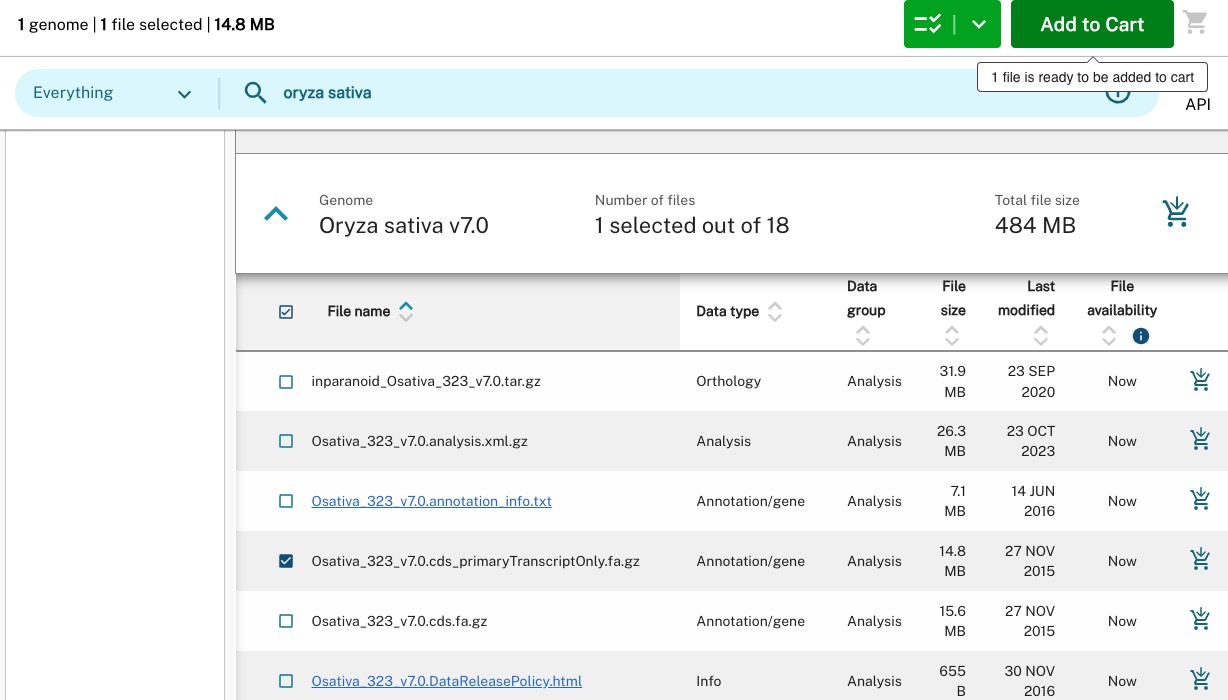
We can then repeat this step for all of the species that we want to download. When we
are ready, we then click the shopping cart icon, and press Download on the next page.
You will have to sign-in or register to download data.
These files can be placed in a folder, where they are now ready to run OrthoFinder.